单细胞分析:细胞聚类(十)
导读
前面我们已经整合了高质量的细胞,现在我们想知道细胞群中存在的不同细胞类型 ,因此下面将进行细胞聚类分析。
学习目标
-
描述评估用于聚类的主成分数量的方法
-
根据重要的主成分对细胞进行聚类
1. 目标
-
生成特定细胞类型的簇并使用已知细胞类型的标记基因来鉴定簇的身份。
-
确定簇是否代表真正的细胞类型或是由于生物学或技术变异而产生的簇,例如细胞周期 S 期的细胞簇、特定批次的簇或具有高线粒体含量的细胞簇。
2. 挑战
-
生物或技术问题可能会导致鉴定出质量差的簇 -
识别每个簇的细胞类型 -
需要保持耐心,因为这可能在聚类和标记基因识别之间进行重复(有时甚至会回到 QC 过滤步骤)
3. 推荐
-
在执行聚类之前,对您对存在的细胞类型有一个很好的了解。了解您是否期望细胞类型复杂性较低或线粒体含量较高,以及细胞是否正在分化。 -
如果您有多个条件的数据,执行整合步骤通常很有帮助。 -
如果需要并且有实验条件,则回归 UMI的数量(默认情况下使用sctransform)、线粒体含量和细胞周期。 -
识别任何无用簇以进行删除或重新进行QC 过滤。无用簇可能包括那些具有高线粒体含量和低 UMI/基因的簇。如果由许多细胞组成,则返回利用 QC 过滤掉,然后重新整合/聚类可能会有所帮助。 -
如果没有将所有细胞类型检测为单独的簇,请尝试更改分辨率或 PC 数量。
4. Set up
在开始之前,创建一个名为 clustering.R 的新脚本。
接下来,让我们加载需要的所有库。
# 单细胞聚类
# 加载包
library(Seurat)
library(tidyverse)
library(RCurl)
library(cowplot)
5. PCs 鉴定
为了克服 scRNA-seq 数据中任何单个基因表达中的广泛技术噪音,Seurat根据从整合的最可变基因的表达中获得的 PCA分数将细胞分配到簇种,每个 PC 基本上代表一个“metagene”,结合相关基因集的信息。因此,确定要在聚类步骤中包含多少 PC 对于确保我们捕获数据集中存在的大部分变异或细胞类型非常重要。
在决定哪些 PC 用于下游聚类分析之前,对 PC 探索很有用。
(a) 探索 PC 的一种方法是使用热图来可视化选定 PC 的最多变异基因,其中基因和细胞按 PCA 分数排序。这里的想法是查看 PC 并确定驱动它们的基因对于区分不同的细胞类型是否有意义。
cells参数指定用于绘图的具有最高负或正 PCA 分数的细胞数。
# 利用热图探索 PCs
DimHeatmap(seurat_integrated,
dims = 1:9,
cells = 500,
balanced = TRUE)
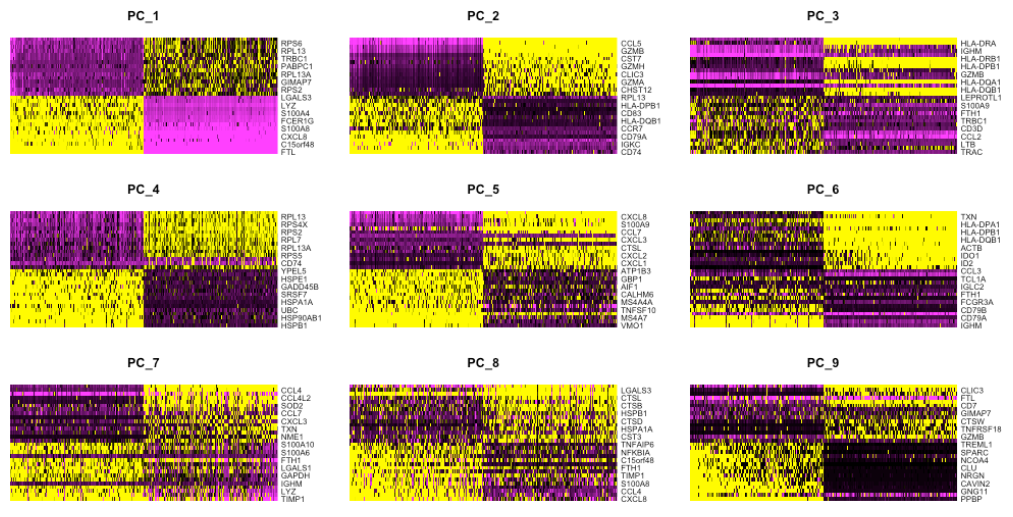
如果我们想探索大量的PC,这种方法可能会很慢并且难以可视化单个基因。同样,为了探索大量 PC,我们可以通过驱动 PC 的 PCA 分数打印出前 10 个(或更多)阳性和阴性基因。
# 打印出驱动 PC 的可变基因
print(x = seurat_integrated[["pca"]],
dims = 1:10,
nfeatures = 5)

(b) elbow图是确定用于聚类 PC 数量的另一种有用方法,以便我们捕获数据中的大部分变化。elbow图可视化了每个 PC 的标准偏差,我们正在寻找标准偏差开始稳定的位置。本质上,elbow出现的位置通常是识别大部分变化的阈值。但是,这种方法可能非常主观。
让我们使用前 40 PCs 绘制elbow图:
# 绘制 elbow 图
ElbowPlot(object = seurat_integrated,
ndims = 40)
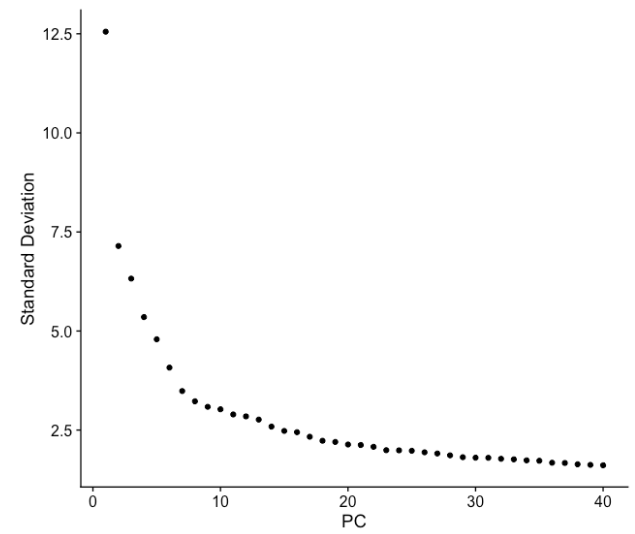
基于此图,我们可以通过elbow出现在 PC8 - PC10 附近的位置粗略确定大部分变化,或者有人可能认为应该是当数据点开始接近 X 轴时,PC为30 左右。这让我们对需要包含的 PC 数量有了一个非常粗略的了解,我们可以以更准确的方式提取此处可视化的信息,这可能更可靠一些。
虽然上述 2 种方法通常与 Seurat 的旧方法一起用于标准化和识别可变基因,但它们不再像以前那样重要。这是因为 SCTransform 方法比旧方法更准确。
-
为什么选择 PC 对旧方法更重要?
较旧的方法将一些变异的技术来源结合到一些较高的 PC 中,因此 PC 的选择更为重要。SCTransform 可以更好地估计方差,并且不会经常在更高的 PC 中包含这些技术变异来源。
理论上,使用 SCTransform,我们选择的 PC 越多,执行聚类时考虑的变化就越多,但是执行聚类需要更长的时间。因此,对于此分析,我们将使用前 40 PCs 来生成细胞簇。
6. 聚类
Seurat 使用基于图的聚类方法,将细胞嵌入到图结构中,使用 K 近邻 (KNN) 图(默认情况下),在具有相似基因表达模式的细胞之间绘制边缘。然后,它试图将该图划分为高度互连的quasi-cliques”或communities.
我们将使用 FindClusters() 函数来执行基于图的聚类。分辨率是设置下游聚类granularity的一个重要参数,需要单独进行优化。对于 3,000 - 5,000 个细胞的数据集,设置在 0.4-1.4 之间的分辨率通常会产生较好的聚类结果。增加的分辨率值会导致更多的簇,这对于更大的数据集通常是必需的。
FindClusters() 函数允许我们输入一系列分辨率,并将计算聚类的granularity。这对于测试哪个分辨率更合适,非常有帮助,而无需为每个分辨率单独运行该函数。
# 确定 K 近邻图
seurat_integrated <- FindNeighbors(object = seurat_integrated,
dims = 1:40)
# 确定各种分辨率的簇
seurat_integrated <- FindClusters(object = seurat_integrated,
resolution = c(0.4, 0.6, 0.8, 1.0, 1.4))
如果我们查看 Seurat 对象的元数据(seurat_integrated@meta.data),则计算出的每个不同分辨率都有一个单独的列。
# 分辨率探索
seurat_integrated@meta.data %>%
View()
开始选择的分辨率,我们通常会选择范围中间的值,例如 0.6 或 0.8。我们将从 0.8 的分辨率开始,使用 Idents() 函数分配簇的标识。
# 分配簇的标识
Idents(object = seurat_integrated) <- "integrated_snn_res.0.8"
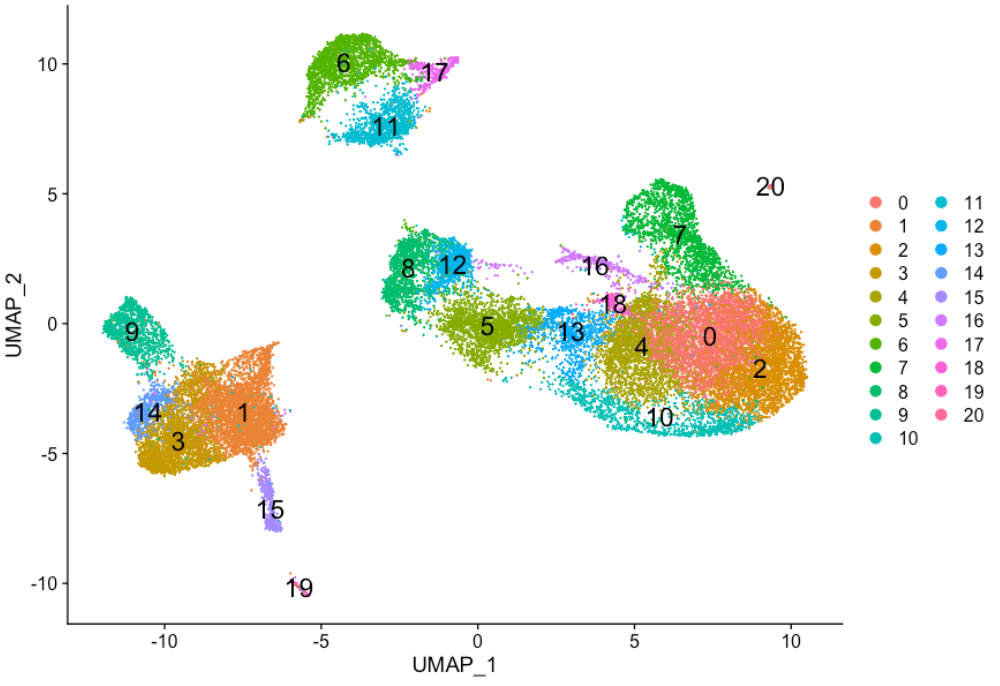
为了可视化细胞簇,有一些不同的降维技术可能会有所帮助。最流行的方法包括 t-SNE 和 UMAP 技术。
这两种方法都旨在将具有相似局部邻域的细胞从高维空间降至低维空间中。这些方法将要求您输入用于可视化的 PCA 维度的数量,我们建议使用相同数量的 PC 作为聚类分析的输入。在这里,我们将继续使用 UMAP 方法来可视化细胞簇。
# UMAP的计算
# seurat_integrated <- RunUMAP(seurat_integrated,
# reduction = "pca",
# dims = 1:40)
# UMAP 可视化
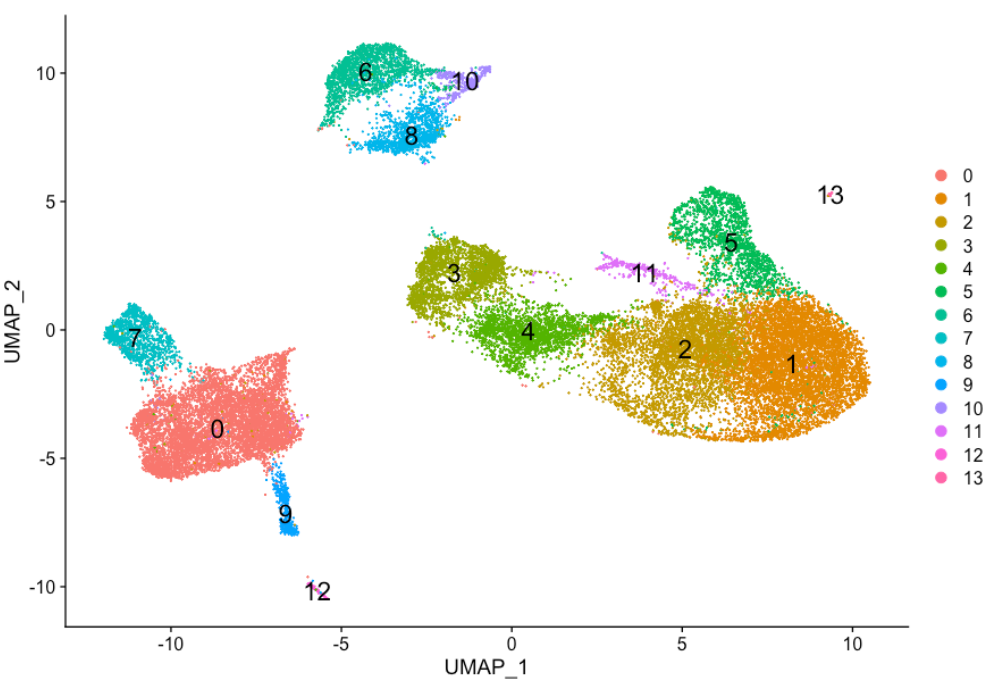
DimPlot(seurat_integrated,
reduction = "umap",
label = TRUE,
label.size = 6)
探索其他分辨率也很有用。它将让您快速了解簇将如何根据分辨率参数发生变化。例如,让我们切换到 0.4 的分辨率:
# 分配簇的标识
Idents(object = seurat_integrated) <- "integrated_snn_res.0.4"
# UMAP 可视化
DimPlot(seurat_integrated,
reduction = "umap",
label = TRUE,
label.size = 6)
欢迎Star -> 学习目录
国内链接 -> 学习目录
本文由 mdnice 多平台发布