生信入门(四)——使用DESeq2进行RNA-seq数据分析
生信入门(四)——使用DESeq2进行RNA-seq数据分析
文章目录
今日学习内容DESeq2分析RNA-seq数据
一、学习目标
- 直观地评估 RNA-seq 数据的质量
- 对 RNA-seq 数据进行基本的差异分析
- 将 RNA-seq 结果与其他实验数据进行比较
- 从 FASTQ 文件中量化转录表达
- 将定量导入 R/Bioconductor
- 执行质量控制和探索性数据分析
- 执行差异表达
- 与其他实验数据重叠
- 构建动态报告
- 整合 DESeq2 和 zinbwave 以获取单细胞 RNA-seq 数据
二、实验数据
1、数据来源
- 本文将展示如何使用替代数据集(tximport小插图中使用的tximportData包)导入 RNA-seq 量化数据。后来我们将加载了数道数据集,其使用计数summarizeOverlaps从GenomicAlignments包。如下所述,我们推荐使用tximport管道来生成计数矩阵,但我们还没有包含气道数据集必要量化文件的 Bioconductor 包。
2、建模计数数据
作为输入,基于计数的统计方法,例如DESeq2 , edgeR , limma with the voom method , DSS、EBSeq 和baySeq期望输入数据是从 RNA-seq 或其他高通量测序实验中获得的计数矩阵。第i行和j 中的值矩阵的第 -th 列表示样本j 中的基因i分配了多少读数(或片段,对于双端 RNA-seq)。类似地,对于其他类型的检测,矩阵的行可能对应于例如结合区域(使用 ChIP-Seq)、细菌种类(使用宏基因组数据集)或肽序列(使用定量质谱)。
3、转录本丰度
在此工作流程中,我们将展示如何使用由Salmon (Patro et al. 2017)软件包量化的转录本丰度。Salmon和其他方法,例如Sailfish 、kallisto 或RSEM ,估计所有(已知的、带注释的)转录本的相对丰度,而没有对齐读取。因为估计转录本的丰度涉及推理步骤,所以估计计数. 大多数方法要么使用称为估计最大化或贝叶斯技术的统计框架来估计丰度和计数。在量化之后,我们将使用tximport (Soneson、Love 和 Robinson 2015)包来组装估计计数和偏移矩阵,以便与 Bioconductor 差异基因表达包一起使用。
将转录本丰度量词与tximport结合使用来生成基因级计数矩阵和归一化偏移的优点是:
- 这种方法纠正了样本中基因长度的任何潜在变化(例如,来自不同异构体的使用)
- 与基于对齐的方法相比,其中一些方法要快得多,需要更少的内存和磁盘使用量
- 可以避免丢弃那些可以与具有同源序列的多个基因对齐的片段(Robert and Watson 2015)。
请注意,转录本丰度量词会跳过存储读取比对(SAM 或 BAM 文件)的大文件的生成,而是生成存储每个转录本的估计丰度、计数和有效长度的较小文件。
salmon量化转录本丰度教程
4、salmon定量
略~因为我看不懂

三、用tximport导入R
1、指定文件位置
量化之后,我们可以使用tximport将数据导入 R 并使用 Bioconductor 包进行统计分析。通常,我们只需将tximport指向quant.sf我们机器上的文件。但是,因为我们将这些文件作为 R 包的一部分进行分发,所以我们必须执行一些额外的步骤,以确定 R 包以及这些文件在您的计算机上的位置。
R 函数system.file可用于找出包中的文件安装在计算机上的哪个位置。这里我们要求提供R 包存储外部数据的目录的完整路径,这是tximportData包的一部分。
BiocManager::install("tximportData")
library("tximportData")
dir<-system.file("extdata",package = "tximportData")
list.files(dir)
list.files(file.path(dir,"salmon"))#量化后的数据在salmon中
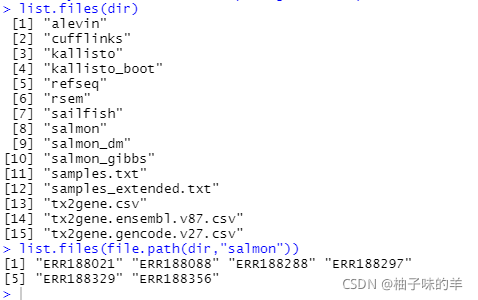
- 此处使用的标识符是来自European Nucleotide Archive的ERR标识符。
我们需要创建一个指向量化文件的命名向量。我们将首先通过读取包含样本 ID 的表来创建文件名向量,然后将其与和结合。(我们对量化文件进行了 gzip 压缩,使数据包更小,这对于我们用于导入文件的 R 函数来说不是问题。)
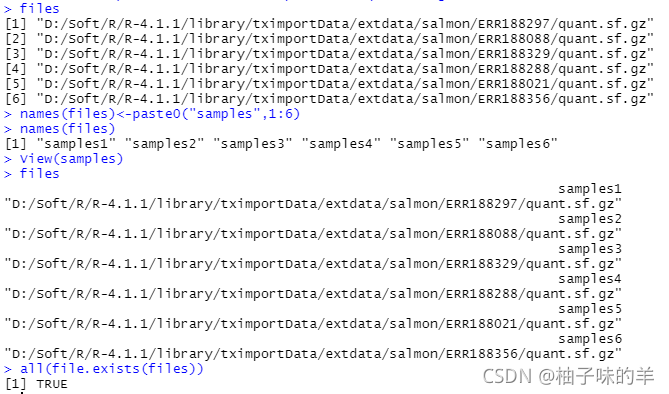
samples<-read.table(file.path(dir,"samples.txt"),header = TRUE)
files<-file.path(dir,"salmon",samples$run,"quant.sf.gz")
names(files)<-paste0("samples",1:6)
all(file.exists(files))
2、将转录本映射到基因
转录本需要与基因 ID 相关联,以便进行基因级汇总。因此,我们将构建一个名为data.frame 的tx2gene两列:1) 转录本 ID 和 2) 基因 ID。列名无关紧要,但必须使用此列顺序。转录本 ID 必须与丰度文件中使用的相同。这可以通过在下载转录组 FASTA 的同时下载tx2geneGTF 文件并使用 Bioconductor 的TxDb基础设施从 GTF 文件生成来最容易地完成。
创建tx2gene data.frame可以通过在TxDb对象上调用AnnotationDbi包中的select函数来完成。以下代码可用于构造这样的表:
#将转录本映射到基因
library("TxDb.Hsapiens.UCSC.hg38.knownGene")
txdb<-TxDb.Hsapiens.UCSC.hg38.knownGene
txdb
k<-keys(txdb,keytype = "TXNAME")
#k 创建tx2gene
tx2gene<-select(txdb,k,"GENEID","TXNAME")
#使用Gencode V27 chr 转录本构建salmon索引
library("readr")
tx2gene<-read_csv(file.path(dir,"tx2gene.gencode.v27.csv"))
head(tx2gene)
3、tximport命令
#tximport 命令——将salmon转录本量化导入R
library("tximport")
library("jsonlite")
txi<-tximport(files,type="salmon",tx2gene = tx2gene)
#txi 对象是一个矩阵列表
names(txi)
txi$counts[1:3,1:3]#提取前三行前三列的txi矩阵中的counts
txi$length[1:3,1:3]#提取前三行前三列的txi矩阵中的length
txi$abundance[1:3,1:3]#提取前三行前三列的txi矩阵中的abundance
txi$countsFromAbundance

如果我们继续使用 GEUVADIS 样本,我们将使用以下代码行创建DESeqDataSet。因为样本之间没有差异(相同的群体和相同的测序批次),我们指定了一个~1的设计公式,这意味着我们只能拟合一个截距项——所以我们不能对这些样本进行差异表达分析。
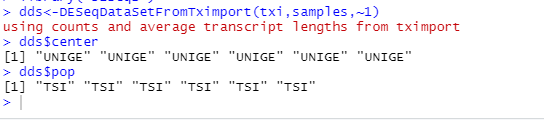
library("DESeq2")
dds<-DESeqDataSetFromTximport(txi,samples,~1)
dds$center
dds$pop
告一段落,明天——>探索性数据分析