Antismash-BigScape次级代谢基因簇鉴定
文章目录
Antismash
概述
antiSMASH - the antibiotics and Secondary Metabolite Analysis SHell,是用来鉴定微生物基因组次级代谢物合成基因簇的软件。
临床上使用的大部分抗生素和药物均来自植物或微生物的天然产物。结合基因组挖掘的经典分离与分析法使得基于基因组的天然产物途径鉴定和描述更为方便。
一般情况下,参与次级代谢途径中生物合成酶基因在基因组上成簇排列,基于指定类型的HMM,antiSMASH数据库能准确鉴定所有已知的次级代谢簇。在antiSMATH中,将次级代谢簇分为24类。
Install
conda create -n antismash -c bioconda antismash
conda activate antismash
(antismash) ok@ubuntu $ download-antismash-databases
Creating checksum of Pfam-A.hmm
PFAM file present and ok for version 32.0
Resfams database present and checked
Creating checksum of TIGRFam.hmm
TIGRFam database present and checked
Creating checksum of proteins.fasta
ClusterBlast fasta file present and checked
Creating checksum of proteins.fasta
ClusterCompare mibig FASTA file present and checked
Pre-building all databases...
done.
# antiSMASH 6.0.1
conda deactivate
# 若出现 Could not remove or rename /home/anaconda3/pkgs/backports_abc-0.5-py_1*,手工删掉,尤其是.conda的文件,手工删掉
# sudo rm -rf /home/anaconda3/pkgs/backports_abc-0.5-py_1*
# manually
## First add the antiSMASH debian repository:
sudo apt-get update
sudo apt-get install -y apt-transport-https
sudo wget http://dl.secondarymetabolites.org/antismash-stretch.list -O /etc/apt/sources.list.d/antismash.list
sudo wget -q -O- http://dl.secondarymetabolites.org/antismash.asc | sudo apt-key add -
sudo apt-get update
## then install the binaries themselves
sudo apt-get install hmmer2 hmmer diamond-aligner fasttree prodigal ncbi-blast+ muscle glimmerhmm
使用
-
Fast run
在没有参数的情况下运行antismash将运行核心检测模块和所有快速的簇特定分析步骤。更多耗时的选项,如ClusterBlast分析、基于簇的PFAM注释、smCoG树生成等将不会被运行。在四核机器上,使用这些选项运行辅酶链霉菌基因组将需要大约两分钟。 -
从prokka注释好的gbk文件预测
(antismash) @ubuntu:/mnt/80G4/TGG_yut/580MAGs_antismash$ time antismash -c 10 --allow-long-headers --output-basename MGGQSC.20.08_metabat2_bin.2 ../580MAGs_prokka/MGGQSC.20.08_metabat2_bin.2.gbk &> MGGQSC.20.08_metabat2_bin.2.log &
#18min
- 从组装好的基因组序列文件预测
(antismash) @ubuntu:/mnt/80G4/TGG_yut/580MAGs_antismash$ time antismash -c 10 --cc-mibig --genefinding-tool prodigal --output-basename MGGQSC.20.08_metabat2_bin.2_genome ../580Batch2_High_quality_MAGs/MGGQSC.20.08_metabat2_bin.2.fa &>MGGQSC.20.08_metabat2_bin.2_genome.log&
# --genefinding-tool 必须要指定
# 20min
# 即使是gbk文件也最好指定--genefinding-tool,因为某些record可能没有CDS区域
nohup time antismash --genefinding-tool prodigal -c 10 --cc-mibig --output-basename GLYI1.20.09_maxbin2_bin.14 /mnt/80G4/TGG_yut/580MAGs_prokka/GLYI1.20.09_maxbin2_bin.14.gbk &
# 注意--output-dir目录必须是空的
- 统计长度
(base) [yutao@myosin antismash_links]$ for i in *gbk;do len=$( head -n1 $i| awk '{print $3}');printf $i"\t"$len"\n";done|sort -k2 -nr >../184OTUs_antismash_BGCs_length.tsv
- 结果:当前目录下生成basename的目录,所以记得在输出目录运行脚本

打开index.html,可以看到2个BGC区域,包括区域、BGC类型以及基因组的位置

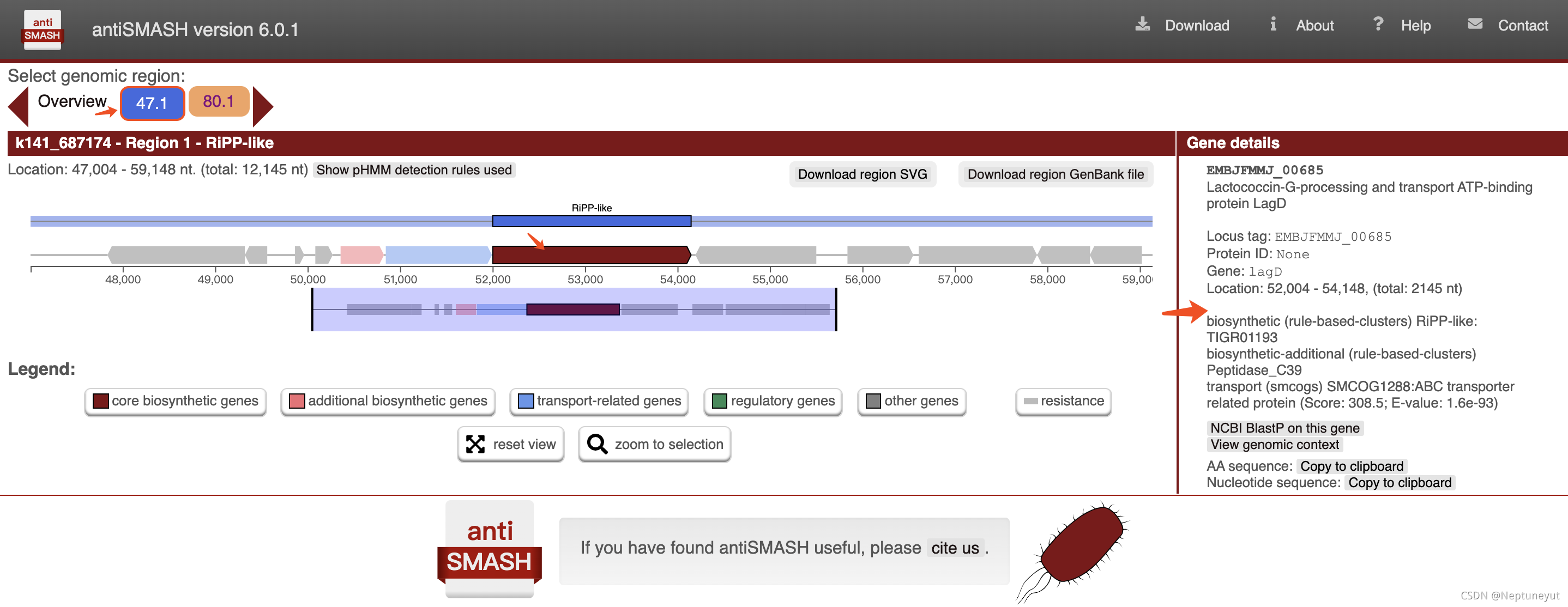
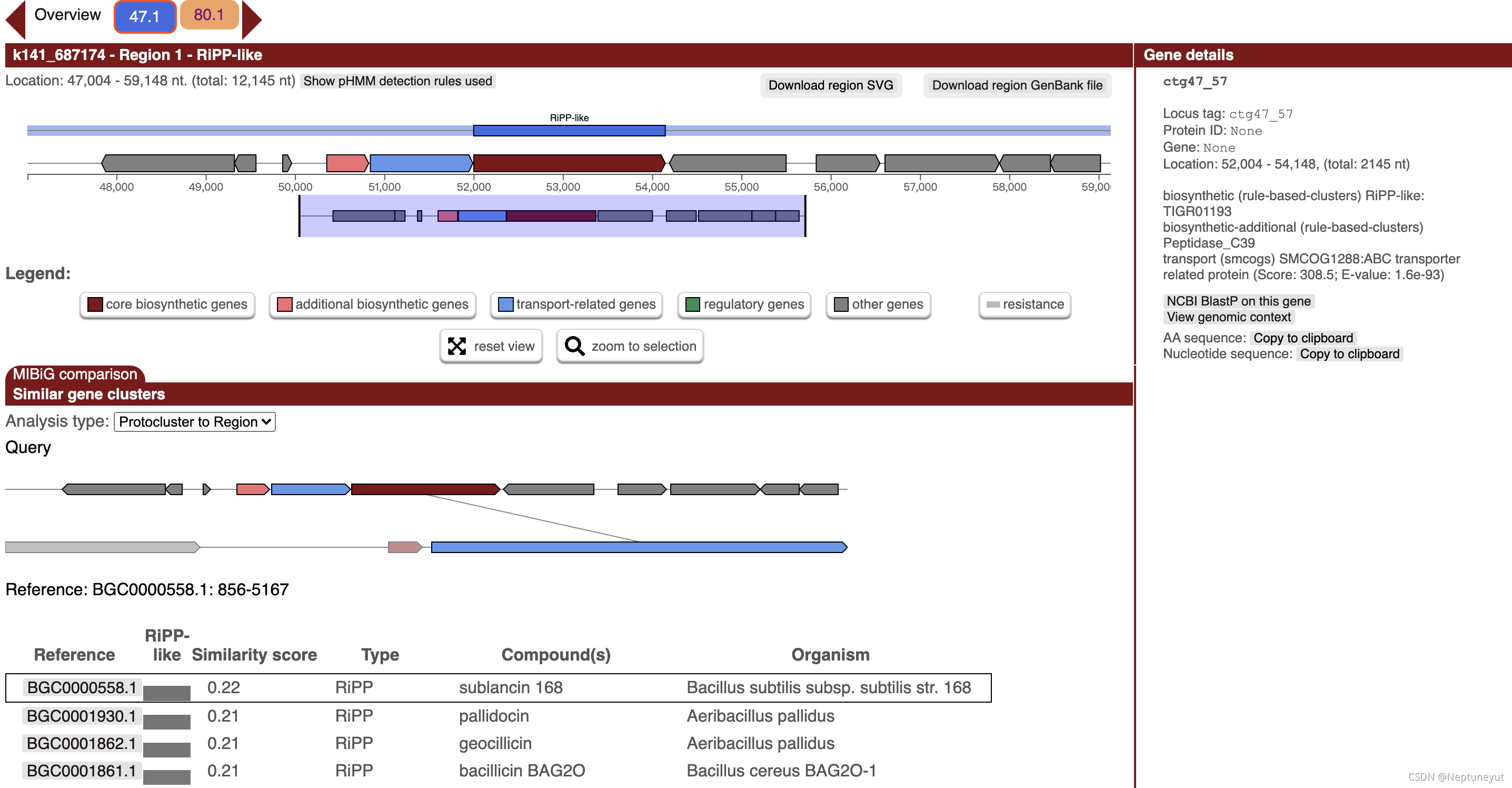
- 如果加上–cc-mibig,则会在最下面展示MiBIG数据库hit到的情况
其他参数
# antiSMASH 6.0.1
--taxon bacteria --cb-general --cb-subclusters --cb-knownclusters --minlength 3000 --hmmdetection-strictness relaxed --genefinding-tool prodigal-m --smcog-trees --clusterhmmer --asf --smcog-trees --pfam2go --allow-long-headers --output-dir . --output-basename test
# --taxon {bacteria,fungi} Taxonomic classification of input sequence. (default: bacteria)
# --cb-general Compare identified clusters against a database of antiSMASH-predicted clusters.
# --cb-subclusters Compare identified clusters against known subclusters responsible for synthesising precursors.
# --cb-knownclusters Compare identified clusters against known gene clusters from the MIBiG database.
# --smcog-trees Generate phylogenetic trees of sec. met. cluster orthologous groups.
# --asf Run active site finder analysis.
# --pfam2go Run Pfam to Gene Ontology mapping module.
# --output-dir OUTPUT_DIR Drectory to write results to.
# --minlength MINLENGTH Only process sequences larger than <minlength> (default: 1000).
# --allow-long-headers Prevents long headers from being renamed
# --output-basename OUTPUT_BASENAME Base filename to use for output files within the output directory.
# --hmmdetection-strictness {strict,relaxed,loose} Defines which level of strictness to use for HMM-based cluster detection, (default: relaxed).
输出详细结果
网络文件
每次运行都会产生自己的一套输出文件,可以使用其他工具进行分析(如Cytoscape)。
Network_Annotations_Full.tsv 一个以制表符分隔的文件,包含输入中成功处理的每个BGC的信息。这包括。BGC名称,来自GenBank文件的原始accesion ID,来自原始GenBank文件的描述,antiSMASH产物预测,BiG-SCAPE类,来自原始GenBank文件的生物体标签,最后,同样来自GenBank文件的分类学字符串。
每个BiG-SCAPE类的文件夹,其中包含。
.网络文件。每个选择的截止点有一个文件。
网络注释文件,包括用于这个特定类别的BGCs。
聚类文件。这些文件包含,对于每个截止点,第一列是BGC名称,第二列用标签隔开,标签代表BGC所在的簇(GCF编号)。
所有这些文件都可以在一个文本编辑器中打开。
GCF and GCC
一旦计算出数据集的距离矩阵,就会对由–cutoffs参数选择的每个截止距离进行基因簇族(Gene Cluster Family, GCF)分配。
BiG-SCAPE输出的交互式可视化将显示数字最大的那一个。
对于每一个截止点,BiG-SCAPE利用所有低于或等于当前截止点的距离创建一个网络。亲和传播聚类算法被应用于这个过程中出现的每个连接组件的子网络。亲和传播的相似性矩阵包括子网络成员之间的所有距离(即它包括那些距离大于当前截止点的成员)。
基因聚类(Gene Cluster Clan, GCC)设置(默认启用)将对GCF进行第二层聚类。为此,亲和传播将再次被应用(即在一个由子连接部件组成的网络上),但网络节点由在参数–clan_cutoff(默认值:0.3)的第一个值中指定的截止水平的GCF代表。聚类将应用于所有GCF的网络,其连接距离低于或等于GCC的截止值(–clan_cutoff参数的第二个值;较大的距离将被丢弃。默认值:0.7)。GCF间的距离被计算为两个家族内BGC间的平均距离。
两个聚类层中使用的亲和传播参数:damping=0.9, max_iter=1000, convergence_iter=200
交互式可视化
通过点击index.html文件或用任何网络浏览器打开该文件来启动互动输出。这个文件位于输出文件夹的根部。
当打开可视化页面时,你会看到一个概览页面。
点击右边的下拉菜单,选择你想可视化的运行。
网页版
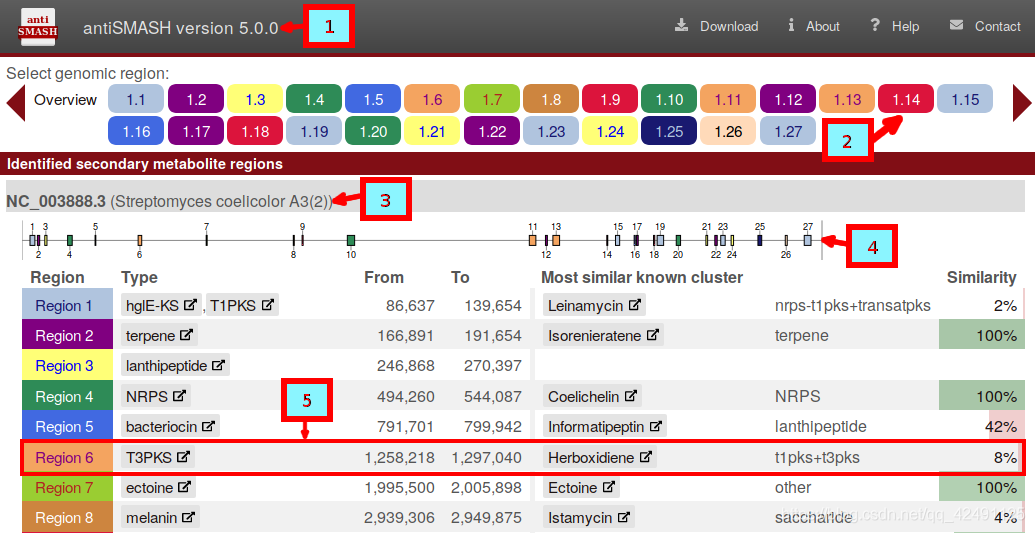
在页面的左上方是antiSMASH的版本信息(1)。antiSMASH结果之间的直接比较应该使用相同的版本以保持一致性,因为不同版本之间的结果可能会发生变化。
在页面的右上方是可能有用的辅助链接。下载允许您下载结果的各个部分。链接到关于antiSMASH的信息(包括出版物)。以及链接到这些文档页面。最后,联系链接到一个有表格的页面,用来发送反馈或问题给antiSMASH开发者。
在顶栏下面是区域按钮(2)。这些按钮在所有antiSMASH HTML页面上都是可见的,并且可以用来快速跳转区域。如果一个区域正在被查看,该区域的匹配按钮将被突出显示。区域按钮的形式是X.Y.,例如,5.7是第五条记录中的第七个区域,区域按钮按预测的次级代谢物类型进行颜色编码。点击 "概述 "按钮,您将回到这个页面。
在这些按钮下是每条记录的摘要,每条记录都以输入文件中给出的记录名称(3)开始。在摘要的顶部是一个图像(4),显示区域在记录中的位置。之后是一个表格,其中有记录中发现的每个区域的摘要,每一行(5)有以下字段:
Region:区域号
Type:antiSMASH检测到的产物类型(点击类型将带你到这些帮助页面中的说明
From,To:该区域的位置(以核苷酸为单位)
Most similar known cluster:MiBIG数据库中最接近的化合物(点击它将带您进入MiBIG条目),以及它的类型(如t1pks+t3pks)。
Similarity:在最接近的已知化合物中,与当前区域内的基因有显著BLAST命中的基因的百分比。
最后两列包含与MiBIG数据库的比较,只有在使用KnownClusterBlast选项运行antiSMASH时才会显示。
点击行中任何有外部链接的地方,将带你进入该区域的详细结果。
- antiSMASH 5区域是如何定义的?
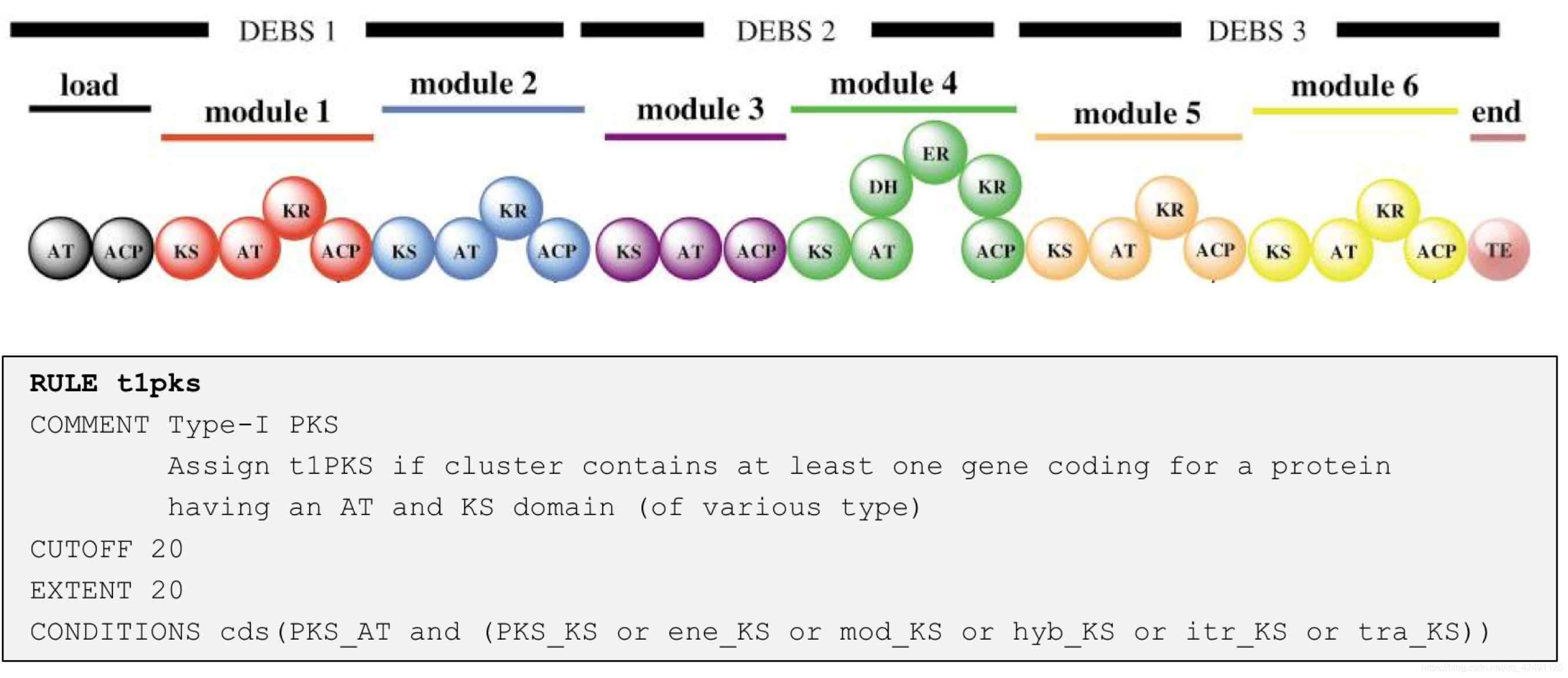
第一步,根据高度保守的酶HMM(隐马尔科夫模型)图谱(核心酶)数据库,对分析序列的所有基因产物进行搜索,这表明了特定的BGC类型。第二步,采用预先定义的簇规则来定义该区域中编码的单个原簇。这些组成了一个核心,由其邻域扩展,邻域由核心上下游编码的基因组成。
下面是一个检测I型PKS的原簇规则的例子,如果一个基因簇至少包含一个包含AT及KS结构域的基因编码区,则会被认为是I型PKS。
其他详细参数
(antismash) @ubuntu:/mnt/80G4/TGG_yut/580MAGs_antismash$ antismash --help
########### antiSMASH 6.0.1 #############
usage: antismash [-h] [options ..] sequence
arguments:
SEQUENCE GenBank/EMBL/FASTA file(s) containing DNA.
--------
Options
--------
-h, --help Show this help text.
--help-showall Show full lists of arguments on this help text.
-c CPUS, --cpus CPUS How many CPUs to use in parallel. (default: 32)
Basic analysis options:
--taxon {bacteria,fungi}
Taxonomic classification of input sequence. (default: bacteria) # 可选择细菌和真菌,是两种不同的预测方式
Additional analysis:
--fullhmmer Run a whole-genome HMMer analysis.
--cassis Motif based prediction of SM gene cluster regions.
--clusterhmmer Run a cluster-limited HMMer analysis.
--tigrfam Annotate clusters using TIGRFam profiles.
--smcog-trees Generate phylogenetic trees of sec. met. cluster orthologous groups.
--tta-threshold TTA_THRESHOLD
Lowest GC content to annotate TTA codons at (default: 0.65).
--cb-general Compare identified clusters against a database of antiSMASH-predicted clusters.
--cb-subclusters Compare identified clusters against known subclusters responsible for synthesising precursors.
--cb-knownclusters Compare identified clusters against known gene clusters from the MIBiG database. # 将预测基因簇与MIBiG数据库已知基因簇比较
--asf Run active site finder analysis.
--pfam2go Run Pfam to Gene Ontology mapping module.
--rre Run RREFinder precision mode on all RiPP gene clusters.
--cc-mibig Run a comparison against the MIBiG dataset
Output options:
--output-dir OUTPUT_DIR
Directory to write results to.
--output-basename OUTPUT_BASENAME
Base filename to use for output files within the output directory.
--html-title HTML_TITLE
Custom title for the HTML output page (default is input filename).
--html-description HTML_DESCRIPTION
Custom description to add to the output.
--html-start-compact Use compact view by default for overview page.
Gene finding options (ignored when ORFs are annotated):
--genefinding-tool {glimmerhmm,prodigal,prodigal-m,none,error}
Specify algorithm used for gene finding: GlimmerHMM, Prodigal, Prodigal Metagenomic/Anonymous mode, or
none. The 'error' option will raise an error if genefinding is attempted. The 'none' option will not run
genefinding. (default: error).
--genefinding-gff3 GFF3_FILE
Specify GFF3 file to extract features from.
BigScape
简介
在生物信息学上,对编码次级代谢产物的生物合成基因组(BGCs)进行挖掘已成为发现自然产品的一个关键策略。在单基因组的基础上,这一过程由antiSMASH等工具来完成。
当研究大量的基因组和元基因组时,必须进行大规模的分析。BiG-SCAPE(生物合成基因相似性聚类和勘探引擎)是一个计算BGC之间距离的工具,以便将BGC多样性映射到序列相似性网络上,然后对其进行处理以自动重建基因簇家族(Gene Cluster Families),即编码高度相似或相同分子的生物合成的基因簇群。BiG-SCAPE对这些相似性网络的交互式可视化可以有效地探索BGCs的多样性,将它们与MIBiG资源库中的参考数据的知识联系起来。
工作原理
BiG-SCAPE(递归)从输入文件夹中读取以GenBank文件形式存储的BGC信息(最好是对应于用antiSMASH等工具识别的基因簇)。
然后,BiG-SCAPE使用Pfam数据库和HMMER套件中的hmmscan来预测每个序列中的Pfam结构域,从而将每个BGC总结为一个Pfam结构域的线性字符串。
对于集合中的每一对BGCs,它们之间的成对距离被计算为Jaccard、Adjacency Index(AI)和Domain Sequence Similarity(DSS)指数的加权组合。产生两种类型的输出:包括网络文件的文本文件和交互式可视化文件。在一次或多次运行中可以考虑不同的距离截止值(即只有原始距离<截止值的配对才被写入最终的.network文件)。
每个截止值的距离将被用来"自动定义"(GCFs和GCCs) “基因簇家族”(GCFs)和 “基因簇家族”(GCCs)。
默认情况下,BiG-SCAPE使用antiSMASH处理的GenBank文件的/产品信息,将分析结果分成八个[BiG-SCAPE类](BiG-SCAPE classes)。每个都有不同的(调整过的)距离成分的[权重](distance indices weights)集合。你也可以选择将所有的BGC类合并成一个网络文件(–mix)并停用默认分类(–no_classify)。也可以通过使用–banned_classes参数来阻止对任何BiG-SCAPE类的分析。
通过 python bigscape.py -h 或进入具体的维基页面,了解更多关于 BiG-SCAPE 选项的信息。
参见维基中的相关页面以获得更详细的信息。
install
Docker engine installation
Install docker engine
BiG-SCAPE and CORASON can run on docker. If you have docker engine installed, please skip this step. This is a Linux minimal docker installation guide, if you do not use Linux or you are looking for a detailed tutorial on Linux/Windows/Mac Docker engine installation please consult Docker: Getting Started.
mkdir ~/bin # not required if you already have that
curl -q https://git.wageningenur.nl/medema-group/BiG-SCAPE/raw/master/run_bigscape > ~/bin/run_bigscape
chmod a+x ~/bin/run_bigscape
$ ~/bin/run_bigscape # download docker image
$ curl -fsSL https://get.docker.com/ | sh
Then type:
$ sudo usermod -aG docker your-user
问题:docker version错误
Got permission denied while trying to connect to the Docker daemon socket at unix:///var/run/docker.sock: Get http://%2Fvar%2Frun%2Fdocker.sock/v1.40/version: dial unix /var/run/docker.sock: connect: permission denied
解决
sudo groupadd docker #添加docker用户组
sudo gpasswd -a $XXX docker #检测当前用户是否已经在docker用户组中,其中XXX为用户名,例如我的,liangll
sudo gpasswd -a $USER docker #将当前用户添加至docker用户组
newgrp docker
Important step:
Log out from your session (restart your machine) and get back in into your user session before the next step. You may need to restart your computer and not just log out from your session in order to changes to take effect.Test your docker engine with the command:
$ docker run hello-world
BiG-SCAPE installation
At the moment, there are two methods available to run BiG-SCAPE:
- Using the pre-built Docker image, a slightly larger download but zero-fuss install on any system that can run Docker.
- Installing all the dependencies (using a virtual environment is recommended).
Installation using Docker
$ mkdir ~/bin # not required if you already have that
$ curl -q https://git.wageningenur.nl/medema-group/BiG-SCAPE/raw/master/run_bigscape > ~/bin/run_bigscape
$ chmod a+x ~/bin/run_bigscape
$ ~/bin/run_bigscape
If BiG-SCAPE is correctly installed the terminal displays the help menu after some seconds:
usage
- 快速使用:
nohup time run_bigscape Streptomyces_BGC-MiBIG Streptomyces_BIGSCAPE_MiBIG -c 30 &>Streptomyces_BIGSCAPE_MiBIG.log&
# -c 线程数
Example dataset
The example dataset consists of three parts: a set of genomes (includes a single cluster from MIBiG), a set of GenBank files (selected clusters predicted from the set of genomes) and the TauD sequence from a Streptomyces genome (NRRL B-1347).
Download and execute the following script to obtain the files:
$ mkdir ~/bin/example # not required if you already have that
$ curl -q https://raw.githubusercontent.com/nselem/bigscape-corason/master/scripts/data_bigscape_corason.sh > ~/bin/example/data_bigscape_corason.sh
$ chmod a+x ~/bin/example/data_bigscape_corason.sh
$ cd ~/bin/example && ~/bin/example/data_bigscape_corason.sh
- dir:The input for BiG-SCAPE is the directory gbks that contains GenBank files of sequences of Biosynthetical gene clusters (BGCs) predicted by antiSMASH. The BiG-SCAPE output will be stored in the directory
example_output.
After BiG-SCAPE has finished successfully, open theindex.htmlfile located inside the ‘example_output’ folder with your browser, (e.g. Chrome or Firefox). The file contains an interactive offline webpage that displays the BiG-SCAPE results and allows you to explore them.
[u@h@bigscape]$ tree gbks/ genomes/
gbks/
├── BGC0000715.1.cluster001.gbk
├── JMGX01000001.1.cluster003.gbk
├── JMGX01000001.1.cluster009.gbk
├── JMQG01000002.1.cluster016.gbk
├── JOBV01000001.1.cluster003.gbk
├── JOBV01000001.1.cluster044.gbk
├── JOBW01000001.1.cluster001.gbk
├── JOBW01000001.1.cluster018.gbk
├── JOBW01000001.1.cluster027.gbk
├── JOES01000001.1.cluster004.gbk
├── JOES01000001.1.cluster049.gbk
├── JOHJ01000001.1.cluster020.gbk
├── JOIW01000001.1.cluster027.gbk
genomes/
├── BGC0000715.1.cluster001.gbk
├── JMGX01.gbk
├── JMQG01.gbk
├── JOBV01.gbk
├── JOBW01.gbk
- bigscape进行BGC大的分类
# 提取*cluster*.gbk文件,将所有cluster的gbk重命名成genome_name+cluster形式并拷贝到一个目录gbk_link中
(antismash) [yutao@myosin C_MIMAG_MAGs_Functions]$ find 184OTU_antismash -iname "*cluster*.gbk" -o -iname "*region*.gbk"|while read i;do a=$(echo $i|awk -F/ '{print $(NF-1)}');b=$(basename $i);cp ${i} 184OTU_BIGSCAPE_out/antismash_links/"${a}-${b}"; done &
# 注意:一定是硬链接或者直接拷贝,软连接会报找不到fasta序列错误
# 另外,基因组和BGC之间作为文件名时,中间不能用特殊字符,例如|~等。
(antismash) [u@h@2661Genomes_Antismash]$ ls -1f */*region*gbk |while read i;do sam=$(echo $i|awk -F/ '{print $1}');id=$(basename $i);cp $i /mnt/nfs/yutao/3241TGG_High_Quality_Genomes/3241Genomes_Antismash/3241Genomes_Antismash_gbks/${sam}~${id};done &
# 或者重命名成sam~bgc的形式,便于后续解析
# 运行bigscape获得BGC分类信息
nohup time run_bigscape gbk_link 1050_Isolates_BIGSCAPE &>log &
注意:BIGSCAPE默认只识别文件名包含‘cluster’以及 'region’的输入文件,所以需要注意输入文件的名称
- BCG分类结果:
/out/network_files/2021-03-08_02-58-20_hybrids_glocal/Network_Annotations_Full.tsv
| BGC | Accesion ID | Description | Product Prediction | BiG-SCAPE class | Organism | Taxonomy |
|---|---|---|---|---|---|---|
| BGC0000715.1.cluster001 | BGC0000715.1 | Spectinomycin biosynthetic gene cluster | amglyccycl | Saccharides | . | |
| JMGX01000001.1.cluster003 | JMGX01000001.1 | Streptomyces rimosus strain R6-500MV9 contig001, whole genome shotgun sequence | nrps | NRPS | . | |
| JMGX01000001.1.cluster009 | JMGX01000001.1 | Streptomyces rimosus strain R6-500MV9 contig001, whole genome shotgun sequence | indole.cf_fatty_acid.nrps | Others | . |
每一行为一个BGC区域:
- 第一列为BGC的GBK文件名
- 第二列该基因簇的id
- 第三列描述了该BGC所在的contig信息,长度和测序深度
- 第四列为BGC的产物名称,例如amglyccycl,表示氨基糖苷/氨基环醇簇
- 第五列则是BiG-SCAPE的大的分类单元:Saccharides糖类化合物,NRPS非核糖体多肽合酶…
获得cluster的长度:
该结果从antismash每个BGCs的gbk文件获得:
# 将所有gbk放到一个目录
[u@h@gbk_link]$ ls|head
102-3_1.cluster001.gbk
102-3_1.cluster002.gbk
102-3_1.cluster003.gbk
# cluster行即为该cluster的长度 :
cluster 1..21929
[u@h@gbk_link]$ head -n15 Y254-3-2-1-1_1.cluster005.gbk
LOCUS 10 21929 bp DNA linear UNK 01-JAN-1980
DEFINITION 10 length=351671 depth=0.67x
ACCESSION 10
VERSION 10
KEYWORDS .
SOURCE
ORGANISM
.
FEATURES Location/Qualifiers
cluster 1..21929
/contig_edge="False"
/cutoff=20000
/extension=10000
/note="Cluster number: 5"
/note="Detection rule(s) for this cluster type: amglyccycl:
# 批量处理(antismash 5.0版本)
[u@h@gbk_link]$awk 'BEGIN{print "Region_filename\tRegion_length"}$0~/cluster.*[0-9]+..[0-9]+/{split($2,a,".");n=split(FILENAME,b,"/");print b[n]"\t"a[3]}' 1778_MAGs_Bigscape/gbk/*gbk >../1778_MAGs_BIGSCAPE_BGC_region_length.tsv
[u@h@gbk_link]$ head ../1050_Isolates_BIGSCAPE_BGC_region_length.tsv
Region_filename Region_length
102-3_1.cluster001.gbk 20819
102-3_1.cluster002.gbk 41089
102-3_1.cluster003.gbk 15513
102-3_1.cluster004.gbk 21896
# antismash version 6.0.1
grep LOCUS *gbk |awk '{OFS="\t";gsub(":LOCUS","",$1);print3,$6}' >../MAGs_Antismash_bgcs_length.tsv &
# 将BGC长度添加,并格式化输出
awk -F"\t" 'BEGIN{OFS="\t";print "GenomeID", "Region_filename", "Region_length", "Product_prediction", "BiG-SCAPE_class"}NR==FNR{sub(/.gbk/,"",$1);gbk[$1]=$1".gbk";len[$1]=$2}NR>FNR && FNR >1{split($1,a,"_");print a[1], gbk[$1], len[$1], $4, $5 }' 1050_Isolates_BIGSCAPE_BGC_region_length.tsv Network_Annotations_Full.tsv
# 可以结合样本信息添加meta信息
awk -F"\t" 'BEGIN{OFS="\t"}NR==1{head=$0}NR==FNR{sub(/ISO_/,"",$2);a[$2]=$0}NR>FNR{if($1=="GenomeID")printf head"\t"$0"\n";if($1 in a)print a[$1],$0}' /mnt/nfs/yutao/Group_info/2661Genome_metadat.tsv 1050_Isolates_BIGSCAPE_BGC_Region_Length.tsv >883Tibetan_Galicer_ISOs_BGCs_info.tsv
- run_bigscape.log
[u@h@test]$ cat log
input 102-3
output bigscape_out
Running BiG-SCAPE
- - Processing input files - -
Including files with one or more of the following strings in their filename: 'cluster', 'region'
Skipping files with one or more of the following strings in their filename: 'final'
Importing GenBank files
Starting with 6 files
Files that had its sequence extracted: 6
Creating output directories
Trying threading on 256 cores
Predicting domains using hmmscan
Predicting domains for 6 fasta files
Finished generating domtable files.
Parsing hmmscan domtable files
Processing 6 domtable files
New domain sequences to be added; cleaning domains folder
Finished generating pfs and pfd files.
Processing domains sequence files
Adding sequences to corresponding domains file
Reading the ordered list of domains from the pfs files
Creating arrower-like figures for each BGC
Parsing hmm file for domain information
Done
Found file with domains colors
Reading BGC information and writing SVG
Finished creating figures
- - Calculating distance matrix - -
Performing multiple alignment of domain sequences
Using hmmalign
launch_hmmalign took 2.677 seconds
Trying to read domain alignments (*.algn files)
Generating distance network files with ALL available input files
Writing the complete Annotations file for the complete set
Working for each BGC class
Sorting the input BGCs
Terpene (2 BGCs)
Writing annotation files
Calculating all pairwise distances
generate_network took 1.153 seconds
Writing output files
Calling Gene Cluster Families
Cutoff: 0.3
PKSother (1 BGCs)
Writing annotation files
Calculating all pairwise distances
generate_network took 2.548 seconds
Others (1 BGCs)
Writing annotation files
Calculating all pairwise distances
generate_network took 4.624 seconds
NRPS (1 BGCs)
Writing annotation files
Calculating all pairwise distances
generate_network took 7.516 seconds
RiPPs (1 BGCs)
Writing annotation files
Calculating all pairwise distances
generate_network took 10.910 seconds
Main function took 61.392 s
- 时间:6101 BGCs耗时1h:08m:05s
输出
输出文件夹结构
cache: 保存分析过程中预先计算的数据。如果 BiG-SCAPE 再次运行并指向同一个输出文件夹,它将尝试读取并重新使用这个目录中的文件。
domains :
对于分析中发现的每个结构域,将生成三个文件。
fasta文件:包含来自所有BGCs的所有蛋白质的同一结构域的序列
stk文件:使用hmmalign以stockholm格式对每个序列进行比对。
algn文件:Fasta文件,包含fasta格式的比对域(从stockholm文件解析而来)。这些是将在DSS中使用的序列。
domtable: 使用hmmscan对每个BGC的蛋白质序列进行域预测的原始输出。
fasta: 每个BGC的蛋白质序列。从GenBank文件中的CDS特征中提取。
pfd: 从domtable文件中解析出的结果,以制表符分隔的格式。这些结果已经被过滤掉了重叠的结构域。列:聚类名称,(每个域)得分,基因ID(如果存在),包络坐标从,包络坐标到(域预测的,以氨基酸为单位),pfam ID,pfam描述符,起始坐标基因,结束坐标基因,内部cds标题。
pfs: 每个BGC文件的预测结构域的列表。
.dict文件:内部文件
html_content:交互式可视化所需的所有代码
logs:目前只保存每次运行中使用的参数(即指向该输出文件夹)和运行时间。
network files:见下一节中的更多信息。
SVG:分析中每个BGC的svg格式的箭头图。每个图都有代表预测域的方框。这些颜色是随机的,但是用户可以通过修改domains_color_file.tsv文件来改变。
结果
- 查看Gene Cluster Family
(gtdbtk) [yutao@globin 2022-10-26_02-24-32_hybrids_glocal]$ for i in NRPS Others PKSI PKS-NRP_Hybrids PKSother RiPPs Terpene;do printf $i"\t"; awk 'NR>1{print $2}' $i/${i}_clustering_c0.30.tsv|sort -u|wc -l;done;echo
NRPS 408
Others 748
PKSI 129
PKS-NRP_Hybrids 38
PKSother 228
RiPPs 805
Terpene 311
# 将所有BGC基因家族文件合并到一起
(gtdbtk) [yutao@globin 2022-10-26_02-24-32_hybrids_glocal]$ cat */*_clustering_c0.30.tsv |grep -v "^#"|
- 可能会出现同一个BGC被并入多个基因家族
$ grep Cluster1_k141_4886715.region001 */*_clustering_c0.30.tsv
NRPS/NRPS_clustering_c0.30.tsv:Cluster1_k141_4886715.region001 1900
PKS-NRP_Hybrids/PKS-NRP_Hybrids_clustering_c0.30.tsv:Cluster1_k141_4886715.region001 663
PKSother/PKSother_clustering_c0.30.tsv:Cluster1_k141_4886715.region001 663
# 查看1对多BGC的情况
$ cat */*_clustering_c0.30.tsv|grep -v "BGC" |sort -u|awk '{print $2}'|sort |uniq -d|wc -l
# 将所有BGC及基因家族对应关系写到文件
$ cat */*_clustering_c0.30.tsv|grep -v "BGC" |sort -u >17629BGCs_bigscape_GCF.tsv
这里的Cluster1_k141_4886715.region001首先被同时纳入到PKSother和PKS-NRP_Hybrids两个大类中,但是基因家族名称都是663,但同时也被纳入NRPS,并且有另外一个家族名称1900
- 网络文件
每次运行都会产生自己的一套输出文件,可以使用其他工具进行分析(如Cytoscape)。
Network_Annotations_Full.tsv 一个以制表符分隔的文件,包含输入中成功处理的每个BGC信息。这包括:BGC名称,来自GenBank文件的原始accesion ID,来自原始GenBank文件的描述,antiSMASH产物预测,BiG-SCAPE类,来自原始GenBank文件的生物体标签,最后,同样来自GenBank文件的分类学字符串。
每个BiG-SCAPE类的文件夹,其中包含。
.network file:每个选择的截止点有一个文件。
每个BiG-SCAPE类的文件夹,其中包含:
网络文件:每个选择的截止点都有一个文件。
The Network Annotation file ,包括用于这个特定类别的BGCs。
The clustering files,这些文件包含,对于每个截止点,第一列是BGC名称,第二列用标签隔开,标签代表BGC所在的簇(GCF编号)。
所有这些文件都可以在一个文本编辑器中打开。
交互式可视化
通过点击index.html文件或用任何网络浏览器打开该文件来启动互动输出。这个文件位于输出文件夹的根部。
当打开可视化页面时,你会看到一个概览页面。
点击右边的下拉菜单,选择你想可视化的运行。
BiG-SCAPE的_input_是目录_gbks_,其中包含由antiSMASH预测的生物合成基因簇(BGCs)序列的GenBank文件。BiG-SCAPE的_输出_将存储在example_output目录下。
在BiG-SCAPE成功完成后,用你的浏览器(如Chrome或Firefox)打开位于’example_output’文件夹内的index.html文件。该文件包含一个互动的离线网页,显示BiG-SCAPE的结果,并允许你探索它们。
要开始探索,在网站顶部选择一个类
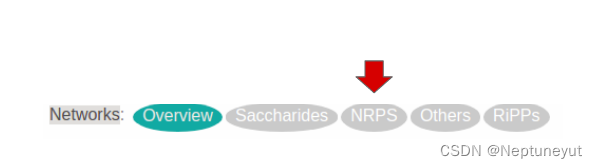
现在,屏幕将显示该类中BGC族的网络可视化。
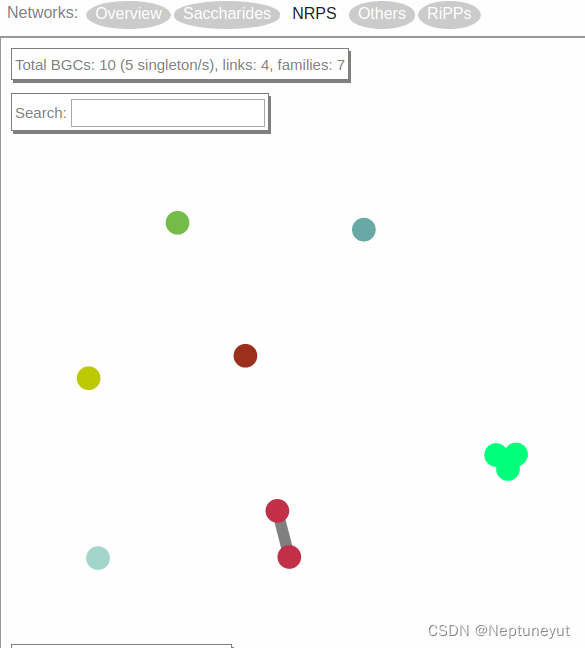
在这个例子下,NRPS类包含10个BGCs,组织在一个有三个成员的基因簇家族中,一个有两个成员的家族和五个单体。
现在在这个网络中选择一个家族,以可视化BGCs的排序,并通过CORASON进行对齐。
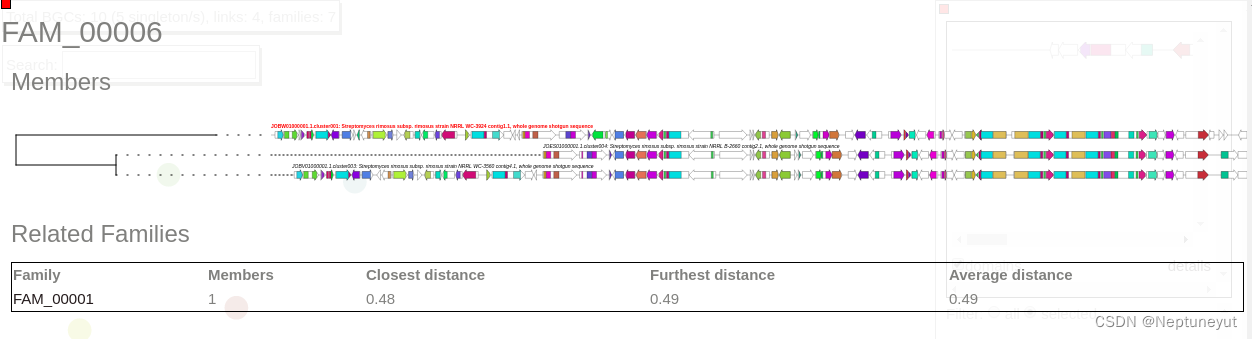
这个家族包含三个成员。
BiG-SCAPE的输出也可以用Cytoscape导入
在另一个例子中,BiG-SCAPE被用来计算103个完整链霉菌基因组中的BGC家族。这个运行的结果可以在这里找到
可能的问题
- 解决antismash分析基因组是出现的genefinding disabled问题
添加如下:
--genefinding-tool prodigal
- header too long and cause unqi id
WARNING 18/05 18:03:00 Fasta header too long: renamed "1807DKMD2|1807DKMD2_Scaff371452" to "c371452_1807DKM.."
WARNING 18/05 18:03:00 Fasta header too long: renamed "1908TGLC1|1908TGLC1_Scaff480182" to "c480182_1908TGL.."
WARNING 18/05 18:03:00 Fasta header too long: renamed "1908TGLC1|1908TGLC1_Scaff480184" to "c480184_1908TGL.."
WARNING 18/05 18:03:00 Fasta header too long: renamed "1908TGLC1|1908TGLC1_Scaff480185" to "c480185_1908TGL.."
WARNING 18/05 18:03:00 Fasta header too long: renamed "1908TGLC2|1908TGLC2_Scaff034255" to "c34255_1908TGL.."
WARNING 18/05 18:03:00 Fasta header too long: renamed "1908TGLC2|1908TGLC2_Scaff034256" to "c34256_1908TGL.."
Traceback (most recent call last):
File "/home//anaconda3/envs/antismash/bin/antismash", line 10, in <module>
sys.exit(entrypoint())
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/__main__.py", line 129, in entrypoint
sys.exit(main(sys.argv[1:]))
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/__main__.py", line 118, in main
antismash.run_antismash(sequence, options)
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/main.py", line 630, in run_antismash
result = _run_antismash(sequence_file, options)
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/main.py", line 683, in _run_antismash
results.records = record_processing.pre_process_sequences(results.records, options,
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/common/record_processing.py", line 358, in pre_proces>
fix_record_name_id(record, all_record_ids, options.allow_long_headers)
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/common/record_processing.py", line 518, in fix_record>
name, _ = generate_unique_id(record.id[:12], all_record_ids, max_length=16)
File "/home//anaconda3/envs/antismash/lib/python3.8/site-packages/antismash/common/record_processing.py", line 573, in generate_u>
raise RuntimeError("Could not generate unique id for %s after %d iterations" % (prefix, counter - start))
RuntimeError: Could not generate unique id for 1908TGLC2|19 after 1000 iterations
因为header id太长,自动截短了名称,导致运行出错,加上--allow-long-headers,允许长名,不会截短
- BIGSCAPE结果少于Antismash
BIGSCAPE通过hmmscan扫描结构与,某些antismash结果扫描不到结构域从而被丢弃。
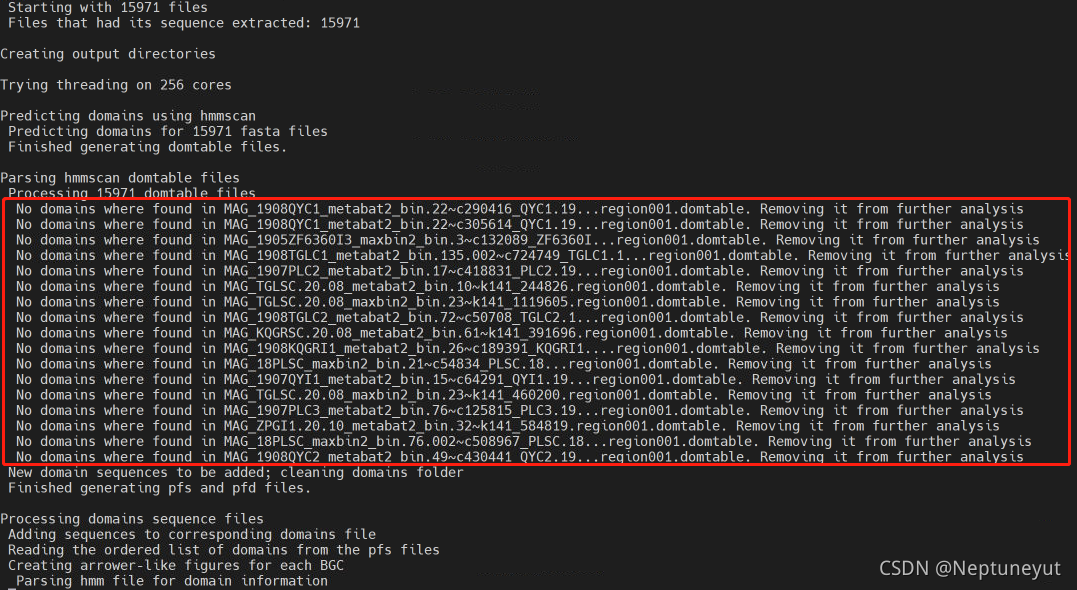
- 内存问题
对于超过10k的BGCs需要注意内存使用,可在运行时指定线程-c 10,但一定要放到命令行最后面,否则默认使用所有核
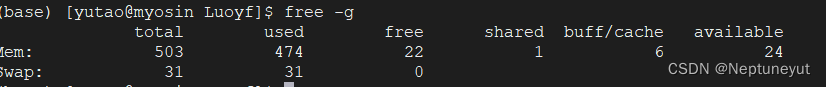
如果不小心使用所有核并且后台一直卡在最后一步使用如下命令杀掉任务:
# 找到运行bigscape的容器
docker ps
# 使用以下命令杀死指定容器(将容器ID或名称替换为实际值):
docker kill <container-id-or-name>